
GEFITINIB MYLAN 250 mg, comprimé pelliculé, boîte de 30 plaquettes thermoformées prédécoupées de 1
Retiré du marché le : 03/02/2023
Dernière révision : 15/06/2022
Taux de TVA : 2.1%
Prix de vente : 754,65 €
Taux remboursement SS : 100%
Base remboursement SS : 754,65 €
Laboratoire exploitant : VIATRIS SANTE
Source :

Géfitinib Mylan est indiqué en monothérapie chez les adultes dans le traitement du cancer bronchique non à petites cellules (CBNPC) localement avancé ou métastatique avec mutations activatrices de l'EGFR-TK (voir rubrique Mises en garde spéciales et précautions d'emploi).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Allaitement (voir rubrique Fertilité, grossesse et allaitement).
Lorsque l'utilisation de Géfitinib Mylan est envisagée en tant que traitement du CBNPC localement avancé ou métastatique, il est important que la mutation de l'EGFR à partir du tissu tumoral soit recherchée pour tous les patients. Si un échantillon de la tumeur n'est pas analysable, de l'ADN tumoral circulant (ADNct) obtenu à partir d'un échantillon de sang (plasma) peut alors être utilisé.
Seul(s) un/des test(s) robuste(s), fiable(s) et sensible(s), ayant démontré son/leur habilité à déterminer le statut de la mutation de l'EGFR au sein de la tumeur ou de l'ADNct, doi(ven)t être utilisé(s) pour éviter les déterminations de faux négatifs ou de faux positifs (voir rubrique Propriétés pharmacodynamiques).
Affections pulmonaires interstitielles (API)
Des affections pulmonaires interstitielles (API), qui peuvent être
aiguës dès le début, ont été observées chez 1,3 % des patients traités
par géfitinib, certains cas ont eu une évolution fatale (voir rubrique Effets indésirables).
Si les patients présentent une aggravation des symptômes respiratoires,
tels que dyspnée, toux et fièvre, le traitement par Géfitinib Mylan
doit être interrompu et le patient doit être examiné rapidement. En cas
de confirmation d'une API, le traitement par Géfitinib Mylan doit être
arrêté et le patient pris en charge de manière appropriée.
Dans une étude pharmaco-épidémiologique japonaise de cas-témoins
portant sur 3 159 patients ayant un CBNPC, sous géfitinib ou
chimiothérapie, et suivis jusqu'à douze semaines, les facteurs de
risque de survenue d'une API suivants (indépendamment du fait que le
patient ait reçu du géfitinib ou une chimiothérapie) ont été identifiés
: tabagisme, indice de performance faible (PS ≥ 2), des preuves
scannographiques de la diminution de la surface pulmonaire saine (≤ 50
%), diagnostic récent de CBNPC (< 6 mois), pneumopathie
interstitielle pré-existante, âge (≥ 55 ans) et pathologie cardiaque
associée. Une augmentation du risque d'API sous géfitinib par rapport à
la chimiothérapie a été principalement observée durant les 4 premières
semaines de traitement (OR ajusté 3,8 ; IC à 95 % compris entre 1,9 à
7,7) ; au-delà le risque relatif était plus faible (OR ajusté 2,5 ; IC
à 95 % compris entre 1,1 à 5,8). Sous géfitinib ou chimiothérapie, le
risque de mortalité chez les patients ayant développé une API est plus
élevé chez ceux qui présentent les facteurs de risque suivants :
tabagisme, preuves scannographiques de la diminution de la surface
pulmonaire saine (≤ 50 %), API pré- existante, âge (≥ 65 ans), et des
zones étendues adhérentes à la plèvre (≥ 50 %).
Hépatotoxicité et insuffisance hépatique
Des anomalies du bilan hépatique (incluant des augmentations de
l'alanine aminotransférase, de l'aspartate aminotransférase, de la
bilirubine) ont été observées, se présentant peu fréquemment sous forme
d'hépatite (voir rubrique Effets indésirables).
Il y a eu des cas isolés d'insuffisance hépatique qui ont, dans
certains cas, conduit à une issue fatale. En conséquence, une
surveillance régulière du bilan hépatique est recommandée. Le géfitinib
devra être utilisé avec prudence en présence de modifications légères à
modérées de la fonction hépatique. L'arrêt du traitement devra être
envisagé en cas de modifications sévères.
Il a été observé qu'une insuffisance hépatique liée à une cirrhose entraînait une augmentation des concentrations plasmatiques de géfitinib (voir rubrique Propriétés pharmacocinétiques).
Interactions avec d'autres médicaments
Les inducteurs du CYP3A4 peuvent augmenter le métabolisme du géfitinib et réduire les concentrations plasmatiques du géfitinib.
Par conséquent, la prise concomitante d'inducteurs du CYP3A4 (par ex.
phénytoïne, carbamazépine, rifampicine, barbituriques ou préparations à
base de plantes contenant du millepertuis/Hypericum perforatum) peut diminuer l'efficacité du traitement et doit être évitée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Chez les patients avec un génotype CYP2D6 métaboliseur lent, un traitement avec un inhibiteur puissant du CYP3A4 peut entraîner une augmentation de la concentration plasmatique du géfitinib. Au début d'un traitement par un inhibiteur du CYP3A4, la survenue d'effets indésirables chez ces patients doit être étroitement surveillée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Une élévation de l'INR (International Normalised Ratio) et/ou des épisodes hémorragiques ont été rapportés chez certains patients traités par la warfarine en association avec le géfitinib (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Chez les patients traités simultanément par la warfarine et le géfitinib, le temps de prothrombine (TP) ou l'INR doivent être régulièrement contrôlés.
Les médicaments qui entraînent une augmentation significative et durable du pH gastrique tels que les inhibiteurs de la pompe à protons et les antagonistes des récepteurs H2 peuvent réduire la biodisponibilité et les concentrations plasmatiques du géfitinib et, par conséquent, en diminuer l'efficacité. Les anti-acides pris régulièrement dans un intervalle de temps rapproché avec l'administration de géfitinib peuvent avoir un effet similaire (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacocinétiques).
Les données des essais cliniques de phase II, avec utilisation concomitante du géfitinib et de vinorelbine, montrent que le géfitinib pourrait augmenter l'effet neutropéniant de la vinorelbine.
Lactose
Géfitinib Mylan contient du lactose. Les patients présentant une
intolérance au galactose, un déficit total en lactase ou un syndrome de
malabsorption du glucose et du galactose (maladies héréditaires rares)
ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d qu'il est essentiellement « sans sodium ».
Précautions d'emploi complémentaires
Il faut recommander aux patients de demander immédiatement un avis
médical s'ils développent une diarrhée sévère ou persistante, des
nausées, des vomissements ou une anorexie qui pourraient indirectement
entraîner une déshydratation. Ces symptômes doivent être pris en charge
selon l'état clinique (voir rubrique Effets indésirables).
Les patients présentant des signes et symptômes aigus ou d'aggravation suggestifs d'une kératite tels que : inflammation de l'œil, larmoiement, sensibilité à la lumière, vision trouble, douleur oculaire et/ou un œil rouge doivent être immédiatement adressés à un ophtalmologiste.
Si un diagnostic de kératite ulcérative est confirmé, le traitement par le géfitinib doit être interrompu, et si les symptômes ne se résolvent pas ou si les symptômes réapparaissent à la réintroduction du géfitinib, un arrêt définitif devra être envisagé.
Dans un essai de phase I/II évaluant l'utilisation du géfitinib et de la radiothérapie dans une population pédiatrique, avec des patients nouvellement diagnostiqués avec une tumeur gliale cérébrale ou une tumeur gliale supratentorielle incomplètement réséquée, 4 cas (1 fatal) d'hémorragie du Système Nerveux Central (SNC) ont été rapportés parmi 45 enfants inclus. Dans le cadre d'un essai clinique avec géfitinib en monothérapie, un autre cas d'hémorragie du SNC a été observé chez un enfant souffrant d'un épendymome. Une augmentation du risque d'hémorragie cérébrale chez les patients adultes présentant un CBNPC et recevant du géfitinib n'a pas été établie.
Des perforations gastro-intestinales ont été rapportées chez des patients prenant du géfitinib. Dans la plupart des cas, elles étaient associées à d'autres facteurs de risque connus, incluant l'administration concomitante de médicaments tels que des stéroïdes ou des AINS, des antécédents d'ulcères gastro- intestinaux, l'âge, le tabagisme ou des métastases intestinales au niveau de la perforation.
Résumé du profil de sécurité d'emploi
Dans les données poolées des études cliniques de phase III ISEL,
INTEREST et IPASS (2 462 patients traités par géfitinib), les effets
indésirables (EI) les plus fréquemment rapportés, survenant chez plus
de 20 % des patients, sont une diarrhée et des réactions cutanées
(incluant éruption cutanée, acné, sécheresse cutanée et prurit). Les
effets indésirables se manifestent habituellement au cours du premier
mois de traitement et sont généralement réversibles.
Environ 8 % des patients ont développé un effet indésirable sévère (Common Toxicity Criteria (CTC), grade 3 ou 4).
Environ 3 % des patients ont arrêté le traitement suite à un effet indésirable.
Des affections pulmonaires interstitielles (API), souvent sévères (CTC grade 3 ou 4), sont survenues chez 1,3 % des patients. Des cas avec évolution fatale ont été rapportés.
Liste tabulée des effets indésirables
Le profil de tolérance présenté dans le tableau 1 est issu du programme
de développement clinique du géfitinib et de l'expérience après
commercialisation. Les effets indésirables ont été classés par
catégorie de fréquence dans le tableau 1 dans la mesure du possible sur
la base de l'incidence d'effets indésirables comparables rapportés dans
la base de données regroupant les essais cliniques de phase III ISEL,
INTEREST et IPASS (2 462 patients traités par géfitinib).
Les fréquences d'apparition des effets indésirables sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (de ≥ 1/100 à < 1/10) ; peu fréquent (de ≥ 1/1 000 à < 1/100) ; rare (de ≥ 1/10 000 à< 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 1 - Réactions indésirables
| Effets indésirables par systèmes d'organes et fréquence | ||
| Troubles du métabolisme et de la nutrition | Très fréquent | Anorexie légère ou modérée (CTC grade 1 ou 2). |
| Affections oculaires | Fréquent | Conjonctivite, blépharite et sécheresse oculaire*, généralement d'intensité légère (CTC grade 1). |
| Peu fréquent | Érosion de la cornée, réversible et parfois en association avec une croissance aberrante des cils. | |
| Kératite (0,12 %) | ||
| Affections vasculaires | Fréquent | Hémorragie, telle qu'épistaxis ou hématurie. |
| Affections respiratoires, thoraciques et médiastinales | Fréquent | Affections pulmonaires interstitielles (1,3 %), parfois grave (CTC grade 3 ou 4). Des cas avec évolution fatale ont été rapportés. |
| Affections gastro- intestinales | Très fréquent | Diarrhée, principalement légère ou modérée (CTC grade 1 ou 2). |
| Vomissements, généralement d'intensité légère (CTC grade 1 ou 2). | ||
| Nausées, généralement d'intensité légère (CTC grade 1). | ||
| Stomatite, essentiellement d'intensité légère (CTC grade 1). | ||
| Fréquent | Déshydratation secondaire à la diarrhée, aux nausées, aux vomissements ou à l'anorexie. | |
| Bouche sèche*, essentiellement d'intensité légère (CTC grade 1). | ||
| Peu fréquent | Pancréatite | |
| Perforation gastro-intestinale | ||
| Affections hépatobiliaires | Très fréquent | Augmentations de l'alanine aminotransférase (ALAT), essentiellement légères à modérées. |
| Fréquent | Augmentations de l'aspartate aminotransférase (ASAT), essentiellement légères à modérées. | |
| Augmentations de la bilirubine totale, essentiellement légère à modérée. | ||
| Peu fréquent | Hépatite** | |
| Affections de la peau et du tissu sous-cutané | Très fréquent | Réactions cutanées, principalement légères ou modérées (CTC grade 1 ou 2), éruption pustuleuse, parfois associées à des démangeaisons et une sécheresse cutanée, des fissures cutanées ou une base érythémateuse. |
| Fréquent | Affections unguéales | |
| Alopécie | ||
| Réactions allergiques (1,1 %), dont l'angioedème et l'urticaire. | ||
| Peu fréquents | Syndrome d'érythrodysesthésie palmo-plantaire | |
| Rare | Eruptions bulleuses incluant nécrolyse épidermique toxique, syndrome de Stevens Johnson et l'érythème polymorphe. | |
| Vasculite cutanée | ||
| Affections du rein et des voies urinaires | Fréquent | Augmentation asymptomatique de la créatininémie |
| | Protéinurie | |
| | Cystite | |
| Rare | Cystite hémorragique | |
| Troubles généraux et anomalies au site d'administration | Très fréquent | Asthénie, principalement légère (CTC grade 1). |
| Fréquent | Fièvre |
* Cet effet indésirable peut se produire en association à d'autres types de sécheresses (principalement des réactions cutanées) observées sous géfétinib.
** Cela inclut des cas isolés d'insuffisance hépatique qui ont, dans certains cas, conduit à une issue fatale.
Affections pulmonaires interstitielles (API)
Dans l'étude INTEREST, l'incidence rapportée des événements de type API
était de 1,4 % (10 patients) dans le groupe géfitinib versus 1,1 % (8
patients) dans le groupe docétaxel. Un événement de type API a eu une
issue fatale chez un patient ayant reçu du géfitinib.
Dans l'étude ISEL l'incidence des événements de type API dans la population globale de l'étude était approximativement de 1 % dans les deux bras de traitement. La majorité des événements de type API a été rapportée chez des patients d'origine asiatique et l'incidence des API chez ces patients recevant du géfitinib ou le placebo était d'environ 3 % et 4 % respectivement. Un événement de type affection pulmonaire interstitielle d'évolution fatale est survenu chez un patient sous placebo.
Dans une étude japonaise de surveillance post-commercialisation (3 350 patients), le taux rapporté des événements de type affection pulmonaire interstitielle était de 5,8 % chez des patients recevant du géfitinib. La proportion des événements de type API avec issue fatale était de 38,6 %.
Dans une étude clinique de phase III en ouvert (IPASS) chez 1 217 patients, comparant le géfitinib à une double chimiothérapie par carboplatine/paclitaxel en première ligne de traitement chez des patients avec un cancer bronchique avancé non à petites cellules en Asie, l'incidence des événements de type API était de 2,6 % dans le bras traité par géfitinib contre 1,4 % dans le bras carboplatine/paclitaxel.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du
médicament est importante. Elle permet une surveillance continue du
rapport bénéfice/risque du médicament. Les professionnels de santé
déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE régulière du bilan hépatique pendant le traitement.
CONTACTER IMMEDIATEMENT LE MEDECIN en cas de :
- Réactions allergiques en particulier si les symptômes incluent un gonflement du visage, des lèvres, de la langue ou de la gorge, des difficultés pour avaler, de l'urticaire et des difficultés pour respirer.
- Essoufflement important ou aggravation soudaine d'un essoufflement, éventuellement avec toux ou fièvre.
- Réactions cutanées sévères s'étendant sur des surfaces importantes du corps, les signes peuvent comprendre rougeur, douleur, ulcération, cloque et décollement de la peau. Les lèvres, le nez, les yeux et les parties génitales peuvent aussi être atteintes.
- Déshydratation suite à une diarrhée sévère ou persistante, des vomissements, des nausées (envie de vomir) ou une perte d'appétit.
-
Troubles oculaires tels que douleur, rougeur, yeux larmoyants,
sensibilité à la lumière, troubles de la vision ou pousse de cils
incarnés.
- Réaction cutanée au niveau des paumes des mains et des plantes des
pieds incluant des picotements, de l'engourdissement, une douleur, un
gonflement ou des rougeurs (connu sous le nom de syndrome
d'érythrodysesthésie palmo-plantaire ou syndrome main-pied).
NE PAS PRENDRE de préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (asthénie).
Femmes en âge de procréer
Les femmes en âge de procréer doivent être incitées à ne pas être enceintes pendant le traitement.
Grossesse
Il n'existe pas de données relatives à l'utilisation du géfitinib chez
la femme enceinte. Les études effectuées chez l'animal ont mis en
évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Le risque potentiel chez l'être humain est inconnu. Géfitinib Mylan ne
doit pas être utilisé pendant la grossesse sauf si indispensable.
Allaitement
Le passage de géfitinib dans le lait maternel est inconnu. Le géfitinib
et ses métabolites s'accumulent dans le lait maternel chez la rate
allaitante (voir rubrique Données de sécurité préclinique). Le
géfitinib est contre-indiqué en cas d'allaitement. De ce fait,
l'allaitement doit être arrêté pendant le traitement par le géfitinib
(voir rubrique Contre-indications).
Le métabolisme du géfitinib se fait via l'isoenzyme CYP3A4 du cytochrome P450 (principalement) et via le CYP2D6.
Substances actives pouvant augmenter les concentrations plasmatiques du géfitinib
Les études in vitro ont montré que le géfitinib est un substrat de la
p-glycoprotéine (P-gp). Les données disponibles ne suggèrent pas
d'impact clinique de ce résultat in vitro.
Les substances qui inhibent le CYP3A4 peuvent diminuer la clairance du géfitinib. L'administration concomitante avec des inhibiteurs puissants de l'activité du CYP3A4 (ex : kétoconazole, posaconazole, voriconazole, inhibiteurs de protéase, clarithromycine, télithromycine) peut augmenter les concentrations plasmatiques du géfitinib. L'augmentation peut avoir un effet clinique significatif puisque les effets indésirables sont liés à la dose et à la durée d'exposition. L'augmentation peut être plus élevée chez les patients avec un génotype métaboliseur lent du CYP2D6. Un traitement préalable avec l'itraconazole (un inhibiteur puissant du CYP3A4) entraîne une augmentation de 80 % de l'aire sous la courbe moyenne du géfitinib chez les volontaires sains. Dans les situations d'un traitement concomitant avec des inhibiteurs puissants du CYP3A4, le patient doit être surveillé étroitement pour les effets indésirables du géfétinib.
Il n'existe aucune donnée concernant un traitement concomitant avec un inhibiteur du CYP2D6, mais les inhibiteurs puissants de cette enzyme peuvent entraîner une augmentation d'environ 2 fois des concentrations plasmatiques du géfitinib chez les métaboliseurs rapides du CYP2D6 (voirrubrique Propriétés pharmacocinétiques). Si un traitement concomitant avec un inhibiteur puissant du CYP2D6 est initié, le patient doit être surveillé étroitement pour les effets indésirables.
Substances actives pouvant diminuer les concentrations plasmatiques du géfitinib
Les substances qui ont une activité inductrice du CYP3A4 peuvent
augmenter le métabolisme et diminuer les concentrations plasmatiques du
géfitinib et, par conséquent, diminuer l'efficacité du géfitinib. Un
traitement concomitant inducteur du CYP3A4 (ex : phénytoïne,
carbamazépine, rifampicine, barbituriques ou millepertuis / Hypericum perforatum)
doit être évité. Un traitement préalable avec la rifampicine (un
inducteur puissant du CYP3A4) a entraîné une réduction de 83 % de
l'aire sous la courbe moyenne chez les volontaires sains (voir rubrique
Mises en garde spéciales et précautions d'emploi).
Les
substances qui entraînent une augmentation significative et durable du
pH gastrique peuvent diminuer les concentrations plasmatiques du
géfitinib et, par conséquent, diminuer l'efficacité du géfitinib. De
fortes doses d'anti-acides à courte durée d'action pourraient entraîner
le même effet en cas de prise régulière dans un intervalle de temps
rapproché avec l'administration de géfitinib.
L'administration concomitante du géfitinib avec la ranitidine à une
dose provoquant une élévation du pH gastrique ≥ 5 a entraîné une
diminution de l'aire sous la courbe moyenne de 47 % du géfitinib chez
les volontaires sains (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Substances actives pouvant avoir leurs concentrations plasmatiques altérées par le géfitinib
Les études in vitro ont montré que le géfitinib a un potentiel
limité à inhiber le CYP2D6. Lors d'une étude clinique réalisée chez des
patients, le géfitinib a été co-administré avec le métoprolol (substrat
du CYP2D6). Cela a résulté en une augmentation de 35 % de l'exposition
au métoprolol. Une telle augmentation pourrait potentiellement être
significative pour les substrats du CYP2D6 avec un index thérapeutique
étroit. Lorsque l'utilisation des substrats du CYP2D6 est envisagée en
association avec le géfitinib, une modification de la dose du substrat
du CYP2D6 doit être envisagée en particulier pour les produits
présentant une fenêtre thérapeutique étroite.
In vitro, le géfitinib inhibe la protéine de transport BCRP, mais la signification clinique de ce résultat est inconnue.
Autres interactions potentielles
Une augmentation de l'INR et/ou des épisodes hémorragiques ont été
décrits chez certains patients prenant de façon concomitante de la
warfarine (voir rubrique Mises en garde spéciales et précautions d'emploi).
Le traitement par Géfitinib Mylan doit être initié et suivi par un médecin expérimenté dans l'utilisation des traitements anticancéreux.
Posologie
La posologie recommandée de Géfitinib Mylan est d'un comprimé de 250 mg
une fois par jour. Si une dose a été oubliée, elle doit être prise
aussitôt que le patient s'en souvient. S'il reste moins de 12 heures
avant la prise de la prochaine dose, le patient ne doit pas prendre la
dose oubliée. Les patients ne doivent pas prendre une double dose (deux
doses en même temps) pour compenser une dose oubliée.
Population pédiatrique
La sécurité et l'efficacité de Géfitinib Mylan chez les enfants et chez
les adolescents âgés de moins de 18 ans n'ont pas été établies. Il
n'existe pas d'utilisation justifiée du géfitinib dans la population
pédiatrique dans l'indication du CBNPC.
Insuffisance hépatique
Les patients avec une insuffisance hépatique modérée à sévère
(Child-Pugh B ou C) suite à une cirrhose présentent une augmentation
des concentrations plasmatiques du géfitinib. Une surveillance étroite
des effets indésirables chez ces patients doit être effectuée. Les
concentrations plasmatiques ne sont pas augmentées chez les patients
qui ont une élévation de l'aspartate aminotransférase (ASAT), des
phosphatases alcalines ou de la bilirubine liées à des métastases
hépatiques (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucun ajustement de dose n'est nécessaire chez les patients ayant une
insuffisance rénale avec une clairance de la créatinine > 20 mL/min.
Des données limitées sont disponibles chez les patients avec une
clairance de la créatinine ≤ 20 mL/min et une attention est requise
chez ces patients (voir rubrique Propriétés pharmacocinétiques).
Patients âgés
Aucun ajustement de dose n'est nécessaire en fonction de l'âge des patients (voir rubrique Propriétés pharmacocinétiques).
CYP2D6 métaboliseurs lents
Aucun ajustement spécifique de dose n'est recommandé chez les patients
avec un génotype CYP2D6 métaboliseurs lents connus, mais une
surveillance étroite des effets indésirables chez ces patients doit
être effectuée (voir rubrique Propriétés pharmacocinétiques).
Ajustement de la dose en fonction de la toxicité
Les patients présentant une diarrhée mal tolérée ou des réactions
cutanées indésirables peuvent être pris en charge efficacement par une
courte interruption du traitement (jusqu'à 14 jours) suivie de sa
reprise à la dose de 250 mg (voir rubrique Effets indésirables).
Pour les patients ne supportant pas le traitement après une
interruption de la thérapie, le géfitinib doit être arrêté et un
traitement alternatif doit être envisagé.
Mode d'administration
Voie orale. Le comprimé peut être pris avec ou sans aliment, chaque jour, approximativement à la même heure.
Le comprimé peut être entièrement avalé avec un peu d'eau ou si
l'administration des comprimés entiers n'est pas possible, les
comprimés peuvent être administrés après dispersion dans l'eau (non
pétillante). Aucune autre boisson ne doit être utilisée.
Sans l'écraser, le comprimé doit être mis dans un demi-verre d'eau. Le
verre doit être remué de temps en temps jusqu'à dissolution du comprimé
(cela peut prendre jusqu'à 20 minutes). La dispersion doit être bue
immédiatement après dissolution totale (c'est-à-dire avant 60 minutes).
Le verre doit être rincé en le remplissant à moitié d'eau qui doit
aussi être bue. La dispersion peut être également administrée par sonde
nasogastrique ou par sonde de gastrostomie.
Durée de conservation :
2 ans
Précautions particulières de conservation :
A conserver à une température ne dépassant pas 30 °C.
Sans objet
Il n'existe pas de traitement spécifique en cas de surdosage du géfitinib. Toutefois, lors d'essais cliniques de phase I, un nombre limité de patients a reçu des doses journalières pouvant atteindre 1 000 mg. Une augmentation de la fréquence et de la sévérité de certains effets indésirables a été observée, essentiellement diarrhée et éruption cutanée. Les effets indésirables associés à un surdosage devront être traités symptomatiquement, en particulier la diarrhée sévère sera prise en charge en fonction des signes cliniques. Dans une étude, un nombre limité de patients ont été traités hebdomadairement avec des doses de 1 500 mg à 3 500 mg. Dans cette étude, l'exposition au géfitinib n'a pas augmenté avec l'augmentation de la dose, les événements indésirables étaient pour la plupart d'intensité légère à modérée et en accord avec le profil de sécurité connu du géfitinib.
Classe pharmacothérapeutique : agents antinéoplasiques, inhibiteurs de protéine kinase, Code ATC : L01EB01
Mécanisme
d'action et effets pharmacodynamiques
Le facteur de
croissance épidermique (EGF) et son récepteur (EGFR [HER1 ; ErbB1]) ont été
identifiés comme des facteurs importants dans le processus de croissance
cellulaire et de prolifération des cellules normales et cancéreuses. La
mutation activatrice de l'EGFR dans une cellule cancéreuse est un facteur
important dans la croissance de la cellule tumorale en bloquant l'apoptose, en
augmentant la production de facteurs angiogéniques et
en facilitant le développement de métastases.
Le géfitinib est une petite molécule inhibitrice sélective de la tyrosine kinase du récepteur du facteur de croissance épidermique et est un traitement efficace pour les patients ayant une tumeur avec mutations activatrices de la tyrosine kinase de l'EGFR, quelle que soit la ligne de traitement. Aucune activité clinique significative n'a été montrée chez les patients ayant une tumeur sans mutation de l'EGFR.
Les mutations activatrices de l'EGFR les plus fréquentes (délétions dans l'exon 19 ; L858R) confèrent des données de réponses robustes étayant la sensibilité au géfitinib ; par exemple un HR de survie sans progression (IC à 95 %) de 0,489 (0,336 ; 0,710) pour le géfitinib par rapport à une double chimiothérapie [WJTOG3405]. Les données de réponse du géfitinib sont plus rares chez les patients dont les tumeurs présentent les mutations les moins fréquentes. Les données disponibles indiquent que les mutations G719X, L861Q et S7681 ont un effet sensibilisant, et que la mutation T790M seule ou des insertions dans l'exon 20 seules sont des mécanismes de résistance.
Résistance
Dans le CBNPC,
la plupart des tumeurs qui présentent des mutations EGFR kinase activatrices
vont développer une résistance au traitement par géfitinib,
avec un temps médian jusqu'à progression de la maladie d'une durée d'un an.
Dans environ 60 % des cas, la résistance est associée à l'apparition d'une
mutation secondaire : T790M pour laquelle un traitement par les ITK EGFR
ciblant cette mutation peut être envisagé comme possibilité de ligne de
traitement suivante. Les autres mécanismes potentiels de résistance qui ont été
rapportés après le traitement par des inhibiteurs de tyrosine kinases de l'EGFR
incluent : contournement du signal, tel que les amplifications de gène HER2 et
MET et les mutations PIK3CA. Un changement phénotypique en cancer du poumon à
petites cellules a aussi été rapporté dans 5 à 10 % des cas.
ADN tumoral
circulant (ADNct)
Dans l'étude
IFUM, le statut de la mutation a été évalué sur des échantillons de tumeur et
d'ADNct obtenus à partir du plasma, en utilisant le
kit TheraScreen EGFR RGQ PCR (Qiagen).
Des échantillons provenant à la fois d'ADNct et de
tumeur ont été analysables pour 652 patients sur les 1 060 sélectionnés. Le
taux de réponse objective (RO) de ces patients porteurs d'une tumeur et
positifs à la mutation ctDNA était de 77 % (IC à 95 %
: 66 % à 86 %) et chez ceux dont seule la tumeur était positive à la mutation,
ce taux était de 60 % (IC à 95 % : 44 % à 74 %).
Tableau 2 - Résumé du statut de mutation initial pour la tumeur et échantillons ctDNA pour tous les patients étudiés, dont les deux échantillons étaient évaluables.
| Mesure | Définition | Taux IFUM % (IC) | IFUM N |
| Sensibilité | Proportion des tumeurs M+ étant M+ par ctDNA | 65,7 (55,8 ; 74,7) | 105 |
| Spécificité | Proportion des tumeurs M étant M par ctDNA) | 99,8 (99,0 ; 100,0) | 547 |
Ces données sont cohérentes avec l'analyse exploratoire prévue dans les sous-groupes de patients Japonais dans l'étude IPASS (Goto 2012). Dans cette étude, l'ADNct obtenu à partir du sérum, et non à partir du plasma, a été utilisé pour l'analyse de la mutation de l'EGFR à l'aide du kit EGFR Mutation Test (DxS) (N = 86). Dans cette étude, la sensibilité était de 43,1 %, et la spécificité de 100 %.
Efficacité
et sécurité cliniques
Première
ligne de traitement
L'étude
clinique randomisée IPASS de phase III en première ligne a été réalisée en
Asie1 chez des patients présentant un CBNPC avancé (stade IIIB ou IV), avec une
histologie de type adénocarcinome, anciens fumeurs légers (arrêt ≥ 15 ans
et ≤ 10 paquets-années) ou n'ayant jamais fumé (voir tableau 3).
1Chine, Hong Kong, Indonésie, Japon, Malaisie, Philippines, Singapour, Taïwan et Thaïlande.
Tableau 3 - Résultats de l'efficacité de l'étude IPASS comparant géfitinib et carboplatine/paclitaxel
| Population | N | Taux de réponse objective et IC à 95 % pour la différence entre les traitementsa | Critère principal, survie sans progression (SSP)a,b | Survie globalea,b | |
| Total | 1 217 | 43,0 % vs 32,2 % [5,3 %, 16,1 %] | HR 0,74 [0,65 ; 0,85] 5,7 m vs 5,8 m p < 0,0001 | HR 0,90 [0,79 ; 1,02] 18,8 m vs 17,4 m p = 0,1087 | |
| EGFR Mutation positive | 261 | 71,2 % vs 47,3 % [12,0 %, 34,9 %] | HR 0,48 [0,36 ; 0,64] 9,5 m vs 6,3 m p < 0,0001 | HR 1,00 [0,76 ; 1,33] 21,6 m vs 21,9 m | |
| EGFR Mutation négative | 176 | 1,1 % vs 23,5 % [-32,5 %, -13,3 %] | HR 2,85 [2,05 ; 3,98] 1,5 m vs 5,5 m p < 0,0001 | HR 1,18 [0,86 ; 1,63] 11,2 m vs 12,7 m | |
| EGFR Mutation inconnue | 780 | 43,3 % vs 29,2 % [7,3 %, 20,6 %] | HR 0,68 [0,58 à 0,81] 6,6 m vs 5,8 m p < 0,0001 | HR 0,82 [0,70 à 0,96] 18,9 m vs. 17,2 m | |
| a | Les données fournies comparent géfitinib à carboplatine/paclitaxel. | ||||
| b | « m » correspond à la médiane en mois. Les nombres entre crochets correspondent à l'intervalle de confiance à 95 % . | ||||
| N | Nombre de patients randomisés. | ||||
| HR | « Hazard ratio » (« hazard ratio » < 1 en faveur de géfitinib) | ||||
Les résultats de qualité de vie diffèrent selon le statut de la mutation de l'EGFR. Chez les patients avec mutation de l'EGFR, les patients traités par géfitinib ont une amélioration significative de la qualité de vie et des symptômes du cancer pulmonaire versus carboplatine/paclitaxel (voir tableau 4).
Tableau 4 - Résultats de qualité de vie de l'étude IPASS comparant géfitinib et carboplatine/paclitaxel
| Population | N | Taux d'amélioration de la QdV FACT La % | Taux d'amélioration des symptômes LCSa % | |
| Total | 1 151 | (48,0 % vs 40,8 %) p = 0,0148 | (51,5 % vs 48,5 %) p = 0,3037 | |
| EGFR Mutation positive | 259 | (70,2 % vs 44,5 %) p < 0,0001 | (75,6 % vs 53,9 %) p = 0,0003 | |
| EGFR Mutation négative | 169 | (14,6 % vs 36,3 %) p = 0,0021 | (20,2 % vs 47,5 %) p = 0,0002 | |
| Les résultats de l'indice des résultats de l'étude confortent les résultats FACT L et LCS. | ||||
| a | Les valeurs présentées correspondent à la comparaison entre géfitinib et carboplatine/paclitaxel. | |||
| N | Nombre de patients évaluables pour les analyses sur la qualité de vie. | |||
| QdV | Qualité de vie | |||
| FACT L | Évaluation fonctionnelle du traitement du cancer du poumon (Functional assessment of cancer therapy lung). | |||
| LSC | Sous-échelle du cancer du poumon (Lung cancer subscale). | |||
Dans l'étude IPASS, le géfitinib a démontré une supériorité sur la SSP, le taux de RO, la QdV et le soulagement symptomatique sans différence significative pour la survie globale par rapport au carboplatine/paclitaxel chez des patients non préalablement traités, présentant un CBNPC localement avancé ou métastatique, dont les tumeurs présentaient des mutations activatrices de la tyrosine kinase de l'EGFR.
Patients
préalablement traités
L'étude
clinique randomisée de phase III INTEREST a été conduite chez des patients
présentant un cancer bronchique non à petites cellules localement avancé ou
métastatique et ayant reçu préalablement une chimiothérapie à base de sels de
platine. Dans la population totale, aucune différence statistiquement
significative n'a été observée entre le géfitinib et
le docétaxel (75 mg/m²) pour la survie globale, la
survie sans progression et le taux de réponse objective (voir tableau 5).
Tableau 5 - Résultats de l'efficacité de l'étude INTEREST comparant géfitinib et docétaxel
| Population | N | Taux de réponse objective et IC à 95 % pour la différence entre les traitementsa | Survie sans progressiona,b | Critère principal, survie globalea,b | |
| Total | 1 466 | 9,1 % vs 7,6 % [•1,5 % ; 4,5 %] | HR 1,04 [0,93 ; 1,18] 2,2 m vs 2,7 m p = 0,4658 | HR 1,020 [0,905 ; 1,150] 7,6 m vs 8,0 m p = 0,7332 | |
| EGFR Mutation positive | 44 | 42,1 % vs 21,1 % [-8,2 %, 46,0 %] | HR 0,16 [0,05 ; 0,49] 7,0 m vs 4,1 m p = 0,0012 | HR 0,83 [0,41 ; 1,67] 14,2 m vs 16,6 m p = 0,6043 | |
| EGFR Mutation négative | 253 | 6,6 % vs 9,8 % [-10,5 %, 4,4 %] | HR 1,24 [0,94 ; 1,64] 1,7 m vs 2,6 m p = 0,1353 | HR 1,02 [0,78 ; 1,33] 6,4 m vs 6,0 m p = 0,9131 | |
| Asiatiquesc | 323 | 19,7 % vs 8,7 % [3,1 %, 19,2 %] | HR 0,83 [0,64 ; 1,08] 2,9 m vs 2,8 m p = 0,1746 | HR 1,04 [0,80 ; 1,35] 10,4 m vs 12,2 m p = 0,7711 | |
| Non asiatiques | 1 143 | 6,2 % vs 7,3 % [•4,3 % ; 2,0 %] | HR 1,12 [0,98 ; 1,28] 2,0 m vs 2,7 m p = 0,1041 | HR 1,01 [0,89 ; 1,14] 6,9 m vs 6,9 m p = 0,9259 | |
| a | Les données fournies comparent géfitinib à docétaxel. | ||||
| b | « m » correspond à la médiane en mois. Les nombres entre crochets correspondent à un intervalle de confiance à 96 % pour le HR de la survie globale pour la population générale, ou un intervalle de confiance à 95 % pour le HR. | ||||
| c | Intervalle de confiance entièrement inférieur à la marge de non infériorité de 1,154 | ||||
| N | Nombre de patients répartis aléatoirement. | ||||
| HR | « Hazard ratio » (« hazard ratio » < 1 en faveur du géfitinib) | ||||
Figures 1 et 2 - Résultats d'efficacité du sous-groupe de patients non asiatiques dans l'étude INTEREST (N de patients = Nombre de patients randomisés)
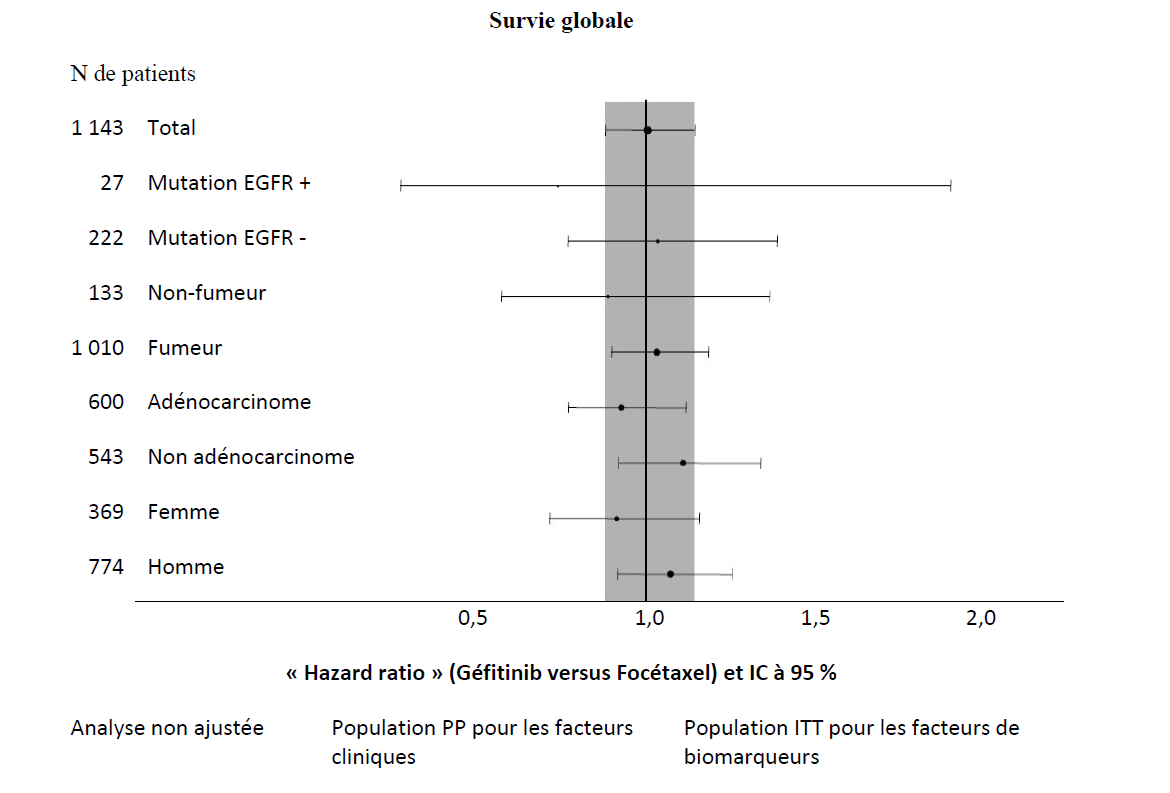
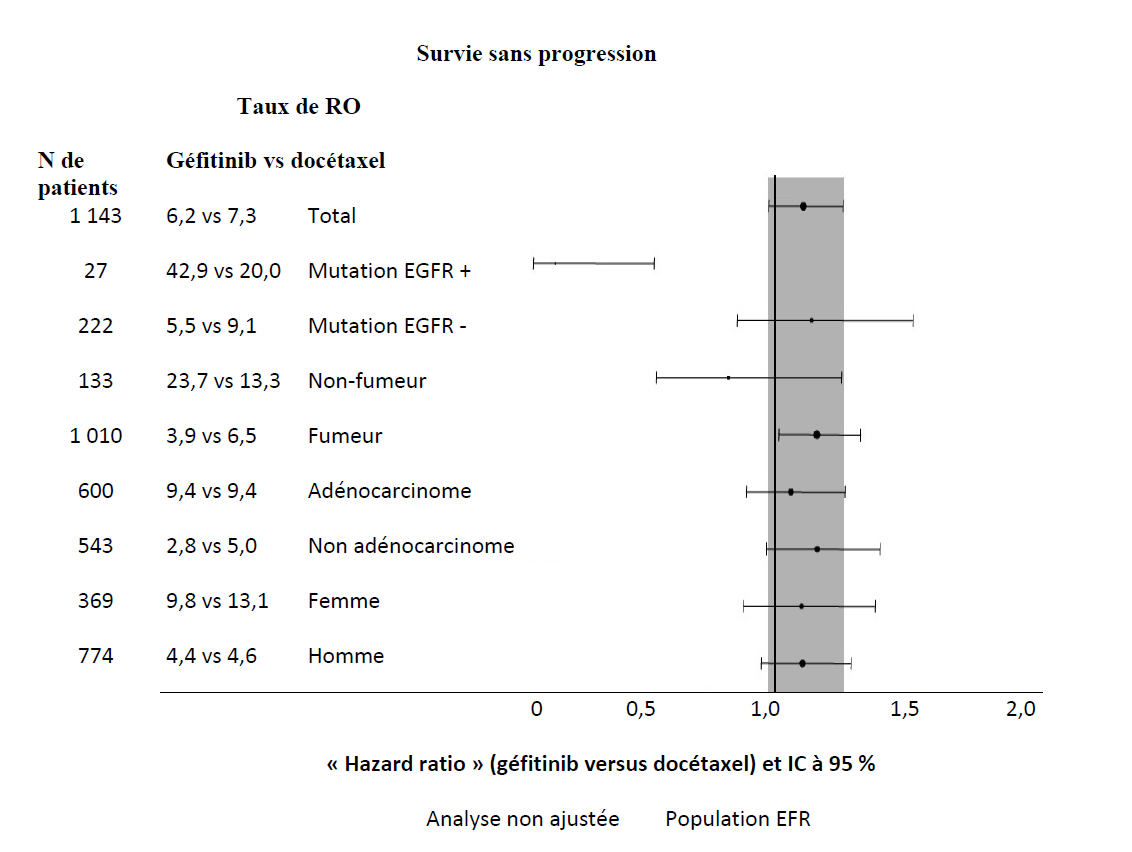
L'étude clinique randomisée de phase III ISEL a été réalisée chez des patients ayant un CBNPC à un stade avancé qui avaient reçu préalablement une ou plusieurs lignes de chimiothérapie et étaient réfractaires ou intolérants à leur dernier traitement. Géfitinib plus traitement symptomatique optimal (Best Supportive Care) a été comparé au placebo plus traitement symptomatique optimal. Le géfitinib n'a pas prolongé la survie dans la population globale. Les résultats de survie diffèrent suivant le statut fumeur et l'origine ethnique (voir tableau 6).
Tableau 6 - Résultats d'efficacité pour le géfitinib et versus placebo dans l'étude ISEL
| Population | N | Taux de réponse objective et IC à 95 % pour la différence entre les traitementsa | Délai de survenue de la rechutea,b | Critère d'évaluation principal, survie globalea,b,c |
| Total | 1 692 | 8,0 % vs 1,3 % [4,7 %, 8,8 %] | HR 0,82 [0,73 ; 0,92] 3,0 m vs 2,6 m p = 0,0006 | HR 0,89 [0,77 ; 1,02] 5,6 m vs 5,1 m p = 0,0871 |
| Mutation EGFR positive | 26 | 37,5 % vs 0 % [-15,1 %, 61,4 %] | HR 0,79 [0,20 ; 3,12] 10,8 m vs 3,8 m p = 0,7382 | HR NC NA vs 4,3 m |
| Mutation EGFR négative | 189 | 2,6 % vs 0 % [•5,6 % ; 7,3 %] | HR 1,10 [0,78 ; 1,56] 2,0 m vs 2,6 m p = 0,5771 | HR 1,16 [0,79 ; 1,72] 3,7 m vs 5,9 m p = 0,4449 |
| Jamais fumeur | 375 | 18,1 % vs 0 % [12,3 %, 24,0 %] | HR 0,55 [0,42 ; 0,72] 5,6 m vs 2,8 m p < 0,0001 | HR 0,67 [0,49 ; 0,92] 8,9 m vs 6,1 m p = 0,0124 | |
| Fumeur | 1 317 | 5,3 % vs 1,6 % [1,4 %, 5,7 %] | HR 0,89 [0,78 ; 1,01] 2,7 m vs 2,6 m p = 0,0707 | HR 0,92 [0,79 ; 1,06] 5,0 m vs 4,9 m p = 0,2420 | |
| Asiatiquesd | 342 | 12,4 % vs 2,1 % [4,0 %, 15,8 %] | HR 0,69 [0,52 ; 0,91] 4,4 m vs 2,2 m p = 0,0084 | HR 0,66 [0,48 ; 0,91] 9,5 m vs 5,5 m p = 0,0100 | |
| Non asiatiques | 1 350 | 6,8 % vs 1,0 % [3,5 %, 7,9 %] | HR 0,86 [0,76 ; 0,98] 2,9 m vs 2,7 m p = 0,0197 | HR 0,92 [0,80 ; 1,07] 5,2 m vs 5,1 m p = 0,2942 | |
| a | Les données fournies comparent géfitinib au placebo. | ||||
| b | « m » correspond à la médiane en mois. Les nombres entre crochets correspondent à l'intervalle de confiance à 95 % . | ||||
| c | Test de log rank stratifié global, sinon modèle à risque proportionnel de Cox. | ||||
| d | L'origine asiatique exclut les patients d'origine indienne et se base sur l'origine ethnique d'un groupe de patients et pas nécessairement au lieu de naissance. | ||||
| N | Nombre de patients randomisés. | ||||
| NC | Non calculé pour le HR de la survie globale, car le nombre d'événements est trop faible. | ||||
| NA | Non atteint | ||||
| HR | « Hazard ratio » (« hazard ratio » < 1 en faveur de géfitinib). | ||||
L'étude IFUM était une étude à bras unique, multicentrique, menée chez des patients caucasiens (n = 106) présentant un CBNPC avec mutations activatrices de l'EGFR, ayant pour but de confirmer que l'activité du géfitinib est similaire dans les populations caucasiennes et asiatiques. Le taux de RO, basé sur l'évaluation de l'investigateur, était de 70 % et la médiane de survie sans progression était de 9,7 mois. Ces résultats sont semblables à ceux rapportés dans l'étude IPASS.
Statut de
la mutation de l'EGFR et caractéristiques cliniques
Les
caractéristiques cliniques du patient telles que le statut Non-fumeur, une
histologie de type adénocarcinome et le sexe féminin, sont des facteurs
prédictifs indépendants de la présence d'une mutation de l'EGFR dans une
analyse multivariée chez 786 patients caucasiens à partir des études réalisées
avec le géfitinib* (voir tableau 7). Les patients
asiatiques ont également une incidence plus élevée de tumeurs ayant une
mutation de l'EGFR.
Tableau 7 - Résumé de l'analyse de régression logistique multivariée pour identifier les facteurs de prédiction indépendants de la présence de mutations de l'EGFR chez 786 patients caucasiens*
| Facteurs de prédiction de la présence de la mutation de l'EGFR | Valeur de p | Risques de mutation de l'EGFR | Valeur de prédiction positive (9,5 % de la population globale est positive à la mutation de l'EGFR [M+]) |
| Tabagisme | < 0,0001 | 6,5 fois plus élevés chez les sujets n'ayant jamais fumé que chez les fumeurs | 28/70 (40 %) des sujets n'ayant jamais fumé sont M+. 47/716 (7 %) des sujets fumeurs sont M+. |
| Histologie | < 0,0001 | 4,4 fois plus élevés pour l'adénocarcinome que pour les autres tumeurs | 63/396 (16 %) des patients ayant une histologie d'adénocarcinome sont M+. 12/390 (3 %) des patients ayant une histologie hors adénocarcinome sont M+. |
| Sexe | 0,0397 | 1,7 fois plus élevés chez les femmes que chez les hommes. | 40/235 (17 %) des femmes sont M+. 35/551 (6 %) des hommes sont M+. |
| *Provenant des études suivantes : INTEREST, ISEL, INTACT 1&2, IDEAL 1&2, INVITE | |||
Absorption
Après administration orale du géfitinib, l'absorption est modérément
lente et le pic des concentrations plasmatiques de géfitinib est
généralement atteint en 3 à 7 heures. La biodisponibilité absolue
moyenne est de 59 % chez les patients cancéreux. La prise de nourriture
est sans effet significatif sur l'exposition au géfitinib. Dans un
essai réalisé auprès de volontaires sains dont le pH gastrique a été
maintenu au-dessus de 5, l'exposition au géfitinib a été réduite de 47
%, vraisemblablement liée à une solubilité réduite du géfitinib dans
l'estomac (voir rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Distribution
Le géfitinib présente un volume de distribution moyen à l'équilibre de
1 400 L, témoignant d'une diffusion tissulaire importante. La fixation
aux protéines plasmatiques est de l'ordre de 90 %. Le géfitinib se lie
à l'albumine sérique et à l'α1-glycoprotéine acide.
Les données in vitro montrent que le géfitinib est un substrat de la protéine de transport membranaire Pg-p.
Biotransformation
Les données in vitro montrent que le CYP3A4 et le CYP2D6 sont
les principaux iso-enzymes du cytochrome P450 impliqué dans le
métabolisme oxydatif du géfitinib.
Les études in vitro ont montré que le géfitinib a un potentiel limité d'inhibition du CYP2D6. Le géfitinib n'a montré chez l'animal aucun effet d'induction enzymatique et aucune inhibition significative de toute autre enzyme du cytochrome P450 (in vitro).
Chez l'homme, le géfitinib est fortement métabolisé. Cinq métabolites ont été complètement identifiés dans les excrétions et 8 métabolites dans le plasma. Le métabolite principal identifié est l'O-desméthyl géfitinib dont le pouvoir inhibiteur de la prolifération cellulaire stimulée par l'EGFR s'avère 14 fois moins puissant que celui du géfitinib. En outre, il n'a pas d'effet inhibiteur sur la croissance de la cellule tumorale chez la souris. Par conséquent, sa contribution à l'activité clinique du géfitinib est donc peu probable.
Il a été montré in vitro que l'O-desméthyl géfitinib est produit via le CYP2D6. Le rôle du CYP2D6 dans la clairance métabolique du géfitinib a été évalué dans un essai clinique chez des volontaires sains génotypés pour le statut CYP2D6. Chez les métaboliseurs lents, il n'a pas été observé de production mesurable d'O-desméthyl géfitinib. Les spectres d'exposition du géfitinib obtenus à la fois chez les métaboliseurs lents et rapides étaient larges et se chevauchaient, mais la moyenne de l'exposition du géfitinib est 2 fois plus élevée dans le groupe des métaboliseurs lents. Les moyennes les plus hautes des expositions susceptibles d'être atteintes chez les patients sans activité du CYP2D6 pourraient être cliniquement significatives, sachant que les effets indésirables dépendent de la dose et de l'exposition.
Élimination
Le géfitinib est principalement excrété sous forme de métabolites dans
les fèces, avec une élimination rénale du géfitinib et de ses
métabolites qui représente moins de 4 % de la dose administrée.
La clairance plasmatique totale du géfitinib est de l'ordre de 500 mL/min et la demi-vie terminale moyenne est de 41 heures chez les patients cancéreux. L'administration de géfitinib une fois par jour se traduit par une accumulation d'un facteur de 2 à 8 avec un état d'équilibre atteint après 7 à 10 doses. À l'état d'équilibre, les concentrations dans le plasma circulant se maintiennent dans les limites d'un facteur de 2 à 3 sur l'intervalle de 24 heures entre deux administrations.
Populations particulières
D'après les analyses de pharmacocinétique de données de population
réalisées chez des patients cancéreux, aucune relation n'a été
identifiée entre la concentration prévisible à l'équilibre et l'âge des
patients, le poids, le sexe, l'origine ethnique ou la clairance de la
créatinine des patients (au-dessus de 20 mL/min).
Insuffisance hépatique
Dans une étude clinique ouverte de phase I à la dose unique de 250 mg
de géfitinib, chez des patients présentant une insuffisance hépatique
légère, modérée ou sévère liée à une cirrhose (suivant la
classification de Child-Pugh), il y a eu une augmentation de
l'exposition dans tous ces groupes par rapport au sujet sain. Chez les
patients présentant une insuffisance hépatique modérée et sévère, une
augmentation moyenne de 3,1 fois de l'exposition au géfitinib a été
observée. Aucun de ces patients n'avait un cancer, tous avaient une
cirrhose et quelques-uns avaient une hépatite. Cette augmentation
pourrait être cliniquement significative sachant que les effets
indésirables dépendent de la dose et de l'exposition au géfitinib.
Le géfitinib a été évalué dans un essai clinique chez 41 patients présentant une tumeur solide, avec une fonction hépatique normale ou une insuffisance hépatique modérée ou sévère (classée selon le grade CTC à l'inclusion pour l'ASAT, les phosphatases alcalines et la bilirubine) due à des métastases hépatiques. Suite à une administration quotidienne de 250 mg de géfitinib, le temps d'atteinte à l'état d'équilibre, la clairance plasmatique totale (CmaxSS) et l'exposition à l'équilibre (ASC24SS) ont été similaires dans les groupes présentant une insuffisance hépatique modérée et une fonction hépatique normale. Les données de 4 patients avec une insuffisance hépatique sévère liée à la présence de métastases hépatiques suggèrent que les expositions à l'état d'équilibre sont également similaires à celles des patients à fonction hépatique normale.
Des cas d'asthénie ont été rapportés au cours du traitement avec le géfitinib. Par conséquent, les patients présentant ce symptôme devront être prudents lors de la conduite des véhicules et l'utilisation des machines.
Les effets
indésirables non observés dans les études cliniques, mais observés chez
l'animal à des doses équivalentes à celles d'une exposition clinique et avec un
impact possible au niveau clinique sont les suivantes :
- Atrophie de
l'épithélium de la cornée et translucidité cornéale
- Nécrose papillaire
rénale
- Nécrose
hépatocellulaire et infiltration des sinusoïdes par des macrophages à
coloration éosinophile.
Les données non cliniques (in vitro) indiquent que le géfitinib a un potentiel d'inhibition du processus de repolarisation du potentiel d'action cardiaque (par exemple l'intervalle QT). L'expérience clinique n'a pas montré de relation causale entre la prolongation du QT et le géfitinib.
Une diminution de la fertilité chez la rate a été observée à la dose de 20 mg/kg/jour.
Des études publiées ont montré que, chez les souris génétiquement modifiées, une absence d'expression de l'EGFR entraîne une anomalie du développement liée à une immaturité épithéliale sur une variété d'organes dont la peau, le tractus gastro-intestinal et les poumons. Lors de l'administration du géfitinib au cours de l'organogénèse, il n'y a eu aucun effet sur le développement embryo-foetal chez le rat à la plus haute dose (30 mg/kg/jour). Cependant chez le lapin, il y a eu une diminution des poids des fœtus à la dose de 20 mg/kg/jour et au-delà. Aucune malformation induite par le géfitinib n'a été rapportée chez les espèces étudiées. Lors de l'administration chez la rate au cours de la gestation et de la mise bas, il y a eu une diminution de la survie de la portée à la dose de 20 mg/kg/ jour.
Après administration orale du géfitinib marqué au C14 à des rates allaitantes 14 jours post-partum, les concentrations de la radioactivité dans le lait étaient 11 à 19 fois plus élevées que dans le sang.
Le géfitinib n'a montré aucun potentiel génotoxique.
Une étude de
carcinogénicité de 2 ans chez le rat a montré une petite, mais significative,
augmentation de l'incidence des adénomes hépatocellulaires chez le rat mâle et
femelle, et des hémangiosarcomes des ganglions lymphatiques mésentériques chez
la rate à la plus haute dose (10 mg/kg/jour) seulement. Les adénomes
hépatocellulaires ont été également observés dans une étude de carcinogénécité
de 2 ans chez la souris. Cette étude a montré une petite augmentation de
l'incidence de ce résultat chez la souris mâle à la demi-dose, et à la fois
chez la souris mâle et femelle à la dose la plus élevée. Ces effets ont atteint
le seuil de significativité chez la souris femelle mais non chez le mâle.
Les doses sans
effet utilisées chez la souris et le rat ne correspondaient pas aux doses
utilisées en clinique. La signification clinique de ces observations est
inconnue.
Les résultats d'une étude in vitro de phototoxicité ont montré que le géfitinib peut avoir un potentiel phototoxique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux spécialistes et services CANCEROLOGIE.
Prescription réservée aux spécialistes et services HEMATOLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux spécialistes et services CANCEROLOGIE.
Prescription réservée aux spécialistes et services HEMATOLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Comprimé pelliculé (comprimé).
Comprimés pelliculés biconvexes, ronds et de couleur marron, d'une dimension d'environ 11,1 mm × 5,6 mm, avec un côté portant la mention « 250 » et un côté vierge.
Plaquette thermoformée en PVC/PVDC/aluminium prédécoupée pour délivrance à l'unité en boîte de 30 ×1 comprimés pelliculés.
Les plaquettes thermoformées peuvent être emballées dans des pochettes en aluminium.
Chaque comprimé pelliculé contient 250 mg de géfitinib.
Excipient à effet notoire :
Chaque comprimé contient 161,0 mg de lactose (sous forme monohydratée).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Lactose monohydraté
Cellulose microcristalline (101)
Crospovidone (type A)
Povidone (K30)
Laurylsulfate de sodium
Stéarate de magnésium
Pelliculage du comprimé
Alcool polyvinylique (E1203)
Macrogol 4000 (E1521)
Talc (E553b)
Dioxyde de titane (E171)
Oxyde de fer rouge (E172)
Oxyde de fer jaune (E172)